Review: Estrogen
The Role of Estrogen in Ovarian Cancer and the Pathways by Which Estrogen Acts
Timothy Ehmann1, Stephanie Barel1, Amitabha Ray2, Daniel Borsch1
1Lake Erie College of Osteopathic Medicine, Seton Hill University, Greensburg, Pennsylvania, 15601
2Alderson Broaddus University, Philippi, West Virginia, 26416, United States
Correspondence: TEhmann08651@med.lecom.edu
Ovarian cancer remains a prevalent and deadly cancer in females. While anti-estrogen therapies have been useful in the treatment of other cancer types, their effectiveness in the treatment of ovarian cancers remains limited due to the limited understanding of estrogen's effects on carcinogenesis and growth promotion in these tissues. This paper aims to summarize the role estrogen plays in ovarian cancer tumorigenesis and its potential value in targeted therapeutics. Estrogen's effects are secondary to interactions with estrogen receptor (ER) α, ER β, and the G protein coupled receptor, GPR30. Genomic signaling of ER-α has been shown to be primarily carcinogenic, while that of ER-β has been shown to be a negative regulator of carcinogenesis. Additionally, ER-α has been found to have carcinogenic effects through non-genomic signaling of the p53, MAPK, EGFR/Her2, PI3K, and IGF/IGFR pathways. GPR30's effects have been found to be more variable and specific to tumor classification. For example, GPR30 is primarily carcinogenic in ovarian epithelial tumor types but appears to have protective effects when highly expressed in granulosa cell tumors. While the above generalizations can be made, a better understanding of estrogen's effects on molecular signaling pathways will potentially allow for development of more effective targeted therapies against ovarian cancer.
Introduction
In the United States, ovarian cancer remains the 5th most common cancer among females today and the second most common gynecological/urinary (GU) cancer among females. Ovarian cancer had an estimated 21750 new cases in 2020 it ranks 4th in mortality among all cancers in females (1,2). Stage I, low grade tumors may be managed with surgery and observation while stage II-IV lesions are typically managed with surgical debulking as well as multiple cycles of chemotherapy; 60% of ovarian cancer cases are stage III on detection (3,4). Chemotherapy, which is typically in the form of platinum-based agents and taxane agents, and surgery still produce a 5 year survival rate of 85-90% for stage I, 57-70% for stage II, 39-59% for stage III, and 17% for stage IV (4). Unfortunately, anti-estrogen treatments are still in their early years with regards to the treatment of ovarian cancer and have shown limited efficacy (5), necessitating a better understanding of this pathway in cancer.
Estrogen interacts with three primary receptors: α, β, and a G protein coupled receptor, GPR30 (17, 26) (Table 1 defines the abbreviations/terms used throughout the paper). These receptors interact with downstream signaling pathways regulating, cell cycle progression, cell division, and migration (10, 37). These pathways may represent a novel target for ovarian cancer treatment, pending a better understanding of their role in carcinogenesis. The treatment of ovarian cancer requires further inroads into the understanding of the pathogenesis of these tumors and the effects of estrogen as it pertains to carcinogenesis and growth promotion. In theory, this will allow for the development of more effective targeted therapies.
Histology of ovarian carcinoma
There are numerous subtypes of ovarian carcinoma, each defined by the cells of origin. Table 2 summarizes some of the major characteristics/statistics regarding each subtype.
Table 1. Definition of terms and abbreviations used throughout the text
| Akt |
Aka Protein kinase B (PKB); A serine/threonine protein kinase which functions in the control of the cell cycle/proliferation |
| Aloesin |
An active component of the aloe vera plant with antiproliferative effects |
| ATF3 |
(Activating transcription factor 3); Member of the ATF/CREB family |
| Bcl-2 |
Anti-apoptotic protein that inhibits release of caspases from within mitochondria |
| β-catenin |
Signaling protein important to the WNT signaling pathway; Involved in cell adhesion as well as transcription/cell proliferation |
| BRAF |
Proto-oncogene; A serine/threonine kinase of the RAF family |
| BTG2 |
(B-Cell Translocation Gene 2); A tumor suppressor of the BTG/TOG gene family |
| Cadherin 6 |
Transmembrane glycoprotein. Aids formation of desmosomes and cell-cell adhesion. |
| CaMKIV |
Calmodulin kinase IV: A calcium-dependent protein kinase |
| Caspase |
Pro-apoptotic enzymes activated when leaked into cytosol from within mitochondria |
| Cathepsin |
A gene promoter activated by ERα and Sp1 |
| c-fos |
Proto-oncogene that produces pro-growth transcription factors |
| c-myc |
Proto-oncogene found on Chromosome 8 |
| CTF-1 |
(CCAAT-binding transcription factor 1); Acts as a DNA-bound cofactor for NF-KB via activation by CaMKIV increasing transcription of p53 |
| CTNNB1 |
Gene that encodes β-catenin |
| Cyclin A |
Protein driving cell cycle progression during the S phase |
| Cyclin B1 |
Protein involved in driving cell cycle progression from G2 to M phase |
| Cyclin D1 |
Protein involved in driving cell cycle progression from G1 to S phase |
| Cyclin E |
Protein involved in driving cell cycle progression from G1 to S phase |
| Delphinidin |
An anthocyanidin; Natural pigment found in berries, red cabbage, grapes, and sweet potatoes with anticarcinogenic effects |
| E-cadherin |
Transmembrane adhesion molecules within belt desmosomes that interacts with actin thereby controlling cell motility, adhesion, and shape |
| EGFR |
(Epidermal growth factor receptor); Induces growth of keratinocytes, fibroblasts, and granulation tissue |
| Transactivation |
Increased expression of EGFR mediated by GPR30 expression |
| G-1 |
(GPER-1-specific compound 1); agonist of GPER1 that inhibits proliferation of cancer cells |
| GRIP1 |
(Glucocorticoid receptor-interacting protein 1); Transcriptional coactivator, SRC family |
| IL-6 |
(Interleukin-6); Cytokine involved in acute systemic inflammation |
| Jun |
Proto-oncogene that produces pro-growth transcription factors |
| MDR-1 |
(Multidrug resistance gene 1); promoter targeted by p53 to repress transcription |
| MEK1/2 |
Protein kinases; Involved in the Ras-Raf-Mek-Erk cascade |
| MMP 11 |
(Matrix metalloproteinase 11); Protein that helps break down the extracellular matrix |
| MMP 17 |
(Matrix metalloproteinase 17); Protein that helps break down the extracellular matrix |
| mTOR |
(Mammalian target of rapamycin); A substrate of Akt that regulates cell metabolism, stimulating protein and lipid synthesis |
| NF-KB |
(Nuclear factor kappa B); Transcription factor involved in secretion of cytokines, DNA transcription, and cell adhesion. |
| p21 |
Cyclin-dependent kinase inhibitor 1 (CDKN1A). Target of p53 leading to cell cycle arrest. |
| p73 |
A tumor suppressor of the p53 family, with strong pro-apoptotic activity to tissues with DNA damage secondary to chemotherapy |
| PTEN |
(Phosphate and tensin homologue) Tumor suppressor gene that inhibits PI3K/Akt signaling |
| RAS |
Proto-oncogene; Family of G proteins involved in signal transduction |
| SDF-1 |
(Stromal cell-derived factor 1); Chemokine promoting autocrine and paracrine cell migration |
| SP-1 |
(Specificity protein 1); Transcription factor that regulates fibulin-1 |
| Src (SRC-3) |
(Steroid hormone receptor coactivator-3) Has the ability to target nuclear hormone receptors as well as act as a transcription factor in other pathways |
| Survivin |
Promoter targeted by p53 to repress transcription; Part of IAP (Inhibitor of apoptosis) family |
| TNFα |
(Tumor necrosis factor α); Inflammatory cytokine |
| TRAF4 |
(Tumor necrosis factor [TNF] receptor-associated factor 4); Target of p53 leading to apoptosis |
Table 2. In vitro and in vivo cell models used for testing of estrogen responsivity
|
ER α |
ER β |
GPR30 |
Unknown |
| + |
— |
+ |
— |
+ |
— |
| Epithelial Cell Tumors |
Serous
Low grade: 87.5% ER α positive1
High grade: 80.7% ER α positive1
49.6% ER β positive82
GPR30 expression high21 |
PEO1
PEO4
PEOR1 |
PEO14
PEO16
41M
59M
OVCAR-3
OVCAR-4
OVCAR-5
CAOV3
OAW42 |
SKOV-3 |
SKOV-3 |
SKOV-3
OVCAR-3 |
|
HEYA8 |
Endometrioid
76.6% ER α positive1
ER β status unknown
GPR30 expression high21 |
|
|
|
|
|
|
|
Mucinous
20.8% ER α positive1
ER β status unknown
GPR30 expression high21 |
|
|
|
|
|
|
|
Clear Cell
19.4% ER α positive1
ER β status unknown
GPR30 status unknown |
|
|
|
|
|
|
A2780
ES-2 |
| Sex Cord Stromal Tumors |
Granulosa Cell
66% ER α positive
53% GPR30 positive20
ER β status unknown |
|
|
|
|
|
|
|
Sertoli/Leydig Cell
79% ER α positive
ER β status unknown
GPR30 status unknown |
|
|
|
|
|
|
|
| Unknown origin |
BG-1 |
|
BG-1 |
|
BG-1 |
|
|
Estrogen Receptors and Carcinogenic Effects in Ovarian Cancer
Estrogen α receptor (ER-α)
The α receptor exists as one of three variants, based on variable splicing of mRNA: ER-α66, ER-α46, and ER-α36 (6, 7). ER-α66 is the primary receptor referenced in the literature. ER-α46 lacks the activating function domain, AF-1,while ER-α36 lacks both AF-1 and AF-2 domains; ER-α46 has been shown to be inhibitory towards ER-α66 in MCF-7 breast cancer cells (6,7). The α receptor (ER-α66) is commonly found in ovarian cancer, but shows a particular predominance in serous carcinomas. In a study performed by Sieh et.al (2013) (8), samples from 2933 females diagnosed with ovarian cancer were examined showing: 87.5% of low-grade serous carcinomas, 80.7% of high grade serous carcinomas, 76.6% of endometrioid carcinomas, 20.8% of mucinous carcinomas, and 19.4% of clear cell carcinomas stained positive for ER (1). In a smaller study of serous and mucinous carcinomas, 60% of these cancers had a ratio of estrogen receptor α:β greater than 1, meaning that cancer cells expressed a significantly greater number of estrogen receptor α molecules when compared to benign ovarian tissues (9). Furthermore, when exposed to estradiol, 5/16 ovarian cancer cell lines all PEO1 and PEO4 cells, staining strongly positive for ER α, showed increased growth compared to ER negative cell lines e.g., PEO14, PEO16, 41M, 59M, OVCAR-3, OVCAR-4, OVCAR-5, A2780, CAOV3 and OAW42 (10). ER positive staining ovarian serous carcinoma PEO4 cells have been shown to proliferate significantly more when exposed to 17-B estradiol; ER negative PEO14 cells displayed no significant response to estradiol (11).
Estrogen receptor β (ER β)
The β receptor appears more frequently in normal ovarian tissue and benign tumors as opposed to malignant tissue/tumor (9, 12). The β receptor has multiple splicing variants: ERB1, ERB2, and ERB5. ERB1 is the primary isoform while ERB2 lacks ligand/DNA binding abilities and serves as a negative regulator of ER α; ERB5 serves a similar inhibitory function towards ER α and ERB1 (12, 13). ERB1 is thought to interfere with ER α activities, inhibit production of ER α, and inhibit cellular proliferation. These effects were exhibited in ER α positive, ER β negative BG-1 cells subsequently infected with adenovirus carrying the ER β gene (14).
Furthermore, ovarian cancer SKOV-3 cells expressing ERB1 exhibited decreased proliferation while caspase activity was found to be increased in ERB1 positive SKOV-3 cell lines compared to ERB1 negative SKOV-3 lines, increasing apoptosis (15). ER β receptor particularly the ERB1 isoform, seems to be protective against malignant evolution/progression. This is further elucidated by He et al, who showed that ER β agonist LY500307 decreased cell viability, promoted tumor suppressor gene expression, and increased apoptotic gene expression (89). Similarly, OSU-ERb-12, a ER β agonist, demonstrated both in vitro and in vivo inhibition of ovarian cell proliferation by inhibiting epithelial to mesenchymal transition, a key transition for malignancies (90). As such, the loss or suppression of ER β appears to be a key to malignant transformation.
GPR30
GPR30 is a membrane bound, G protein coupled receptor, sensitive to estradiol. The GPR30 receptor mediates signaling through multiple mechanisms (16, 17). The primary role of GPR30 in ovarian carcinogenesis is disputed currently. In BG-1 ovarian cancer cells, GPR30 was shown to activate the c-fos gene independently of ER α activity, suggesting that GPR30 may have effects on carcinogenesis independent of the estrogen receptor (17).
In ovarian cysts, higher levels of GPR30 mRNA were found in malignant lesions compared to benign, correlating with increased tumor size, advanced stage, and increased metastatic ability (18). Smith et. al (2009) showed that GPR30 was present more frequently in ovarian carcinoma than in lower risk ovarian tumors and correlated with a lower survival rate (19).
In granulosa cell tumors, GPR30 was found to be present in 53% of samples and correlated with poor survival in newly diagnosed cases (20). Comparison of 42 samples of various benign, borderline, and malignant epithelial ovarian carcinoma samples revealed that malignant samples were more likely to have increased amounts of GPR30 (21). However, GPR30 was shown to exhibit an anti-proliferative role in SKOV-3 and OVCAR-3 ovarian cancer cell lines. In these cells, GPR30 was found to mediate cell cycle arrest and inhibit cell proliferation (22).
Molecular Signaling Pathways
Estrogen receptors contain a ligand domain, a DNA binding domain, as well as domains for the binding and interaction with coactivators and corepressors (23). When estrogen enters the cell, it binds these receptors, inducing a conformational change and allowing for binding to estrogen response elements (ERE) within DNA as well as transcriptions factors (24, 25). Estrogen receptors can localize to the cytosol and nucleus, both initiated via a common pathway (26, 27). Cytosolic receptors, after binding estrogen, do not necessarily localize to the nucleus but rather associate with various other cytosolic proteins, affecting intracellular signaling cascades. ER activity can be divided into genomic and non-genomic, with interplay between the two groups (28).
Genomic activities of ER
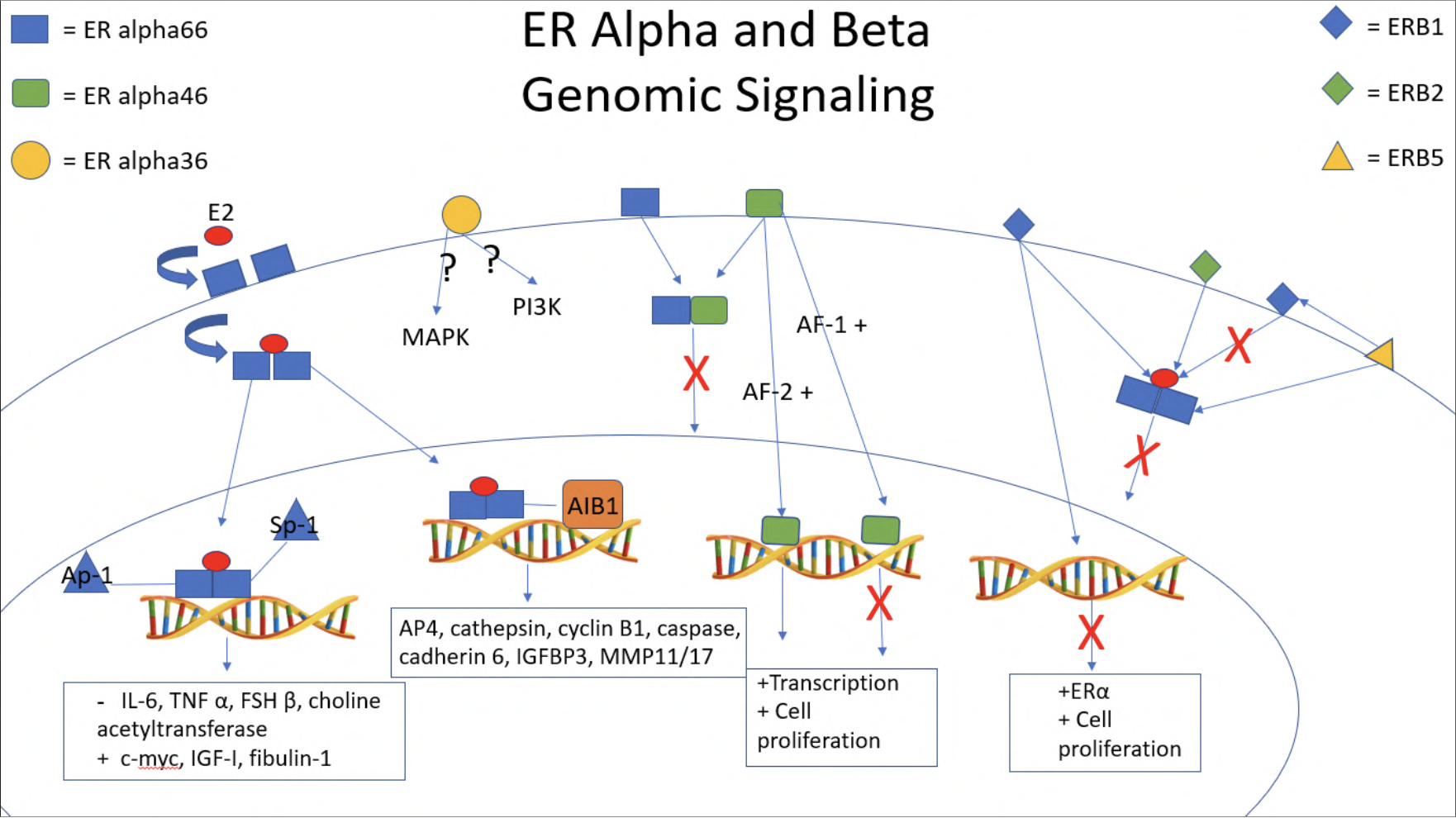
Genomic signaling includes the activation of transcription factors and coactivators or corepressors via estrogen receptors α and β, as well as the direct binding of the ER to ERE regions within DNA. In ovarian cancer cells, genes directly regulated by estradiol through ERE regions include the genes for: AP4 DNA binding protein, cathepsin, cyclin B1, caspase 4, IGFBP3, cadherin 6, matrix metalloproteinases 11 and 17, in PEO1 cells, (10) and stromal cell-derived factor 1 (SDF-1) in BG-1 cells (29), as shown in Figure 1.
Furthermore, a large family of p160 coactivators have been found to mediate ER/estrogen mediated genomic actions. This includes AIB1/SRC-3 and GRIP1 (24, 30). AIB1 (SRC-3) has been shown to be overexpressed in BG-1 ovarian cancer cells. These same AIB1 overexpressing cells have shown increased transcription activity when treated with estradiol, eluding to AIB1's role as a coactivator for ER (31). AIB1 is present in 68.7% of epithelial ovarian cancer samples with a significantly higher expression in cancerous tissue (32) and correlates with worse overall survival in epithelial cancer patients as well as higher grade tumors (33).
AP1 and Sp1 are two highly important transcription factors regulated by the estrogen receptors. Estrogen receptor α has been shown to bind and activate the AP1 complex, which requires intact activating function domains, AF-1 and AF-2. Interestingly, the estrogen β receptor lacks an AF-1 domain and as such only interacts with AP1 when treated with anti-estrogens, which normally block estrogen-α receptor mediated effects (30). Estradiol has been shown to have an inhibitory effect on the AP1 transcriptional pathway, suppressing expression of IL-6, TNF α, FSH β, and choline acetyltransferase (34) as shown in Figure 1. However, in CAOV-3, OVCAR-3, and A2780-ER ovarian cancer cells expressing ER α, estradiolwas found to induce the expression of c-myc and IGF-I through an ER/estradiol/AP1 binding complex, suggesting a gene dependent ER mechanism (35), shown in Figure 1. Furthermore, components of AP-1, fos and jun proteins, have been found in elevated levels within ovarian tumors (36). Fibulin-1, a protein that plays a role in migration and motility of cells, has been shown to be overexpressed in BG-1, PEO4, and OVCAR-3 ovarian cancer cells and is responsive to estradiol treatment through ER α mediation (37). The fibulin-1 promoter was subsequently found to have 2 Sp1 binding sites, both required for estradiol/ER mediated fibulin transcription (37).
P53: Cross-talk between genomic and non-genomic
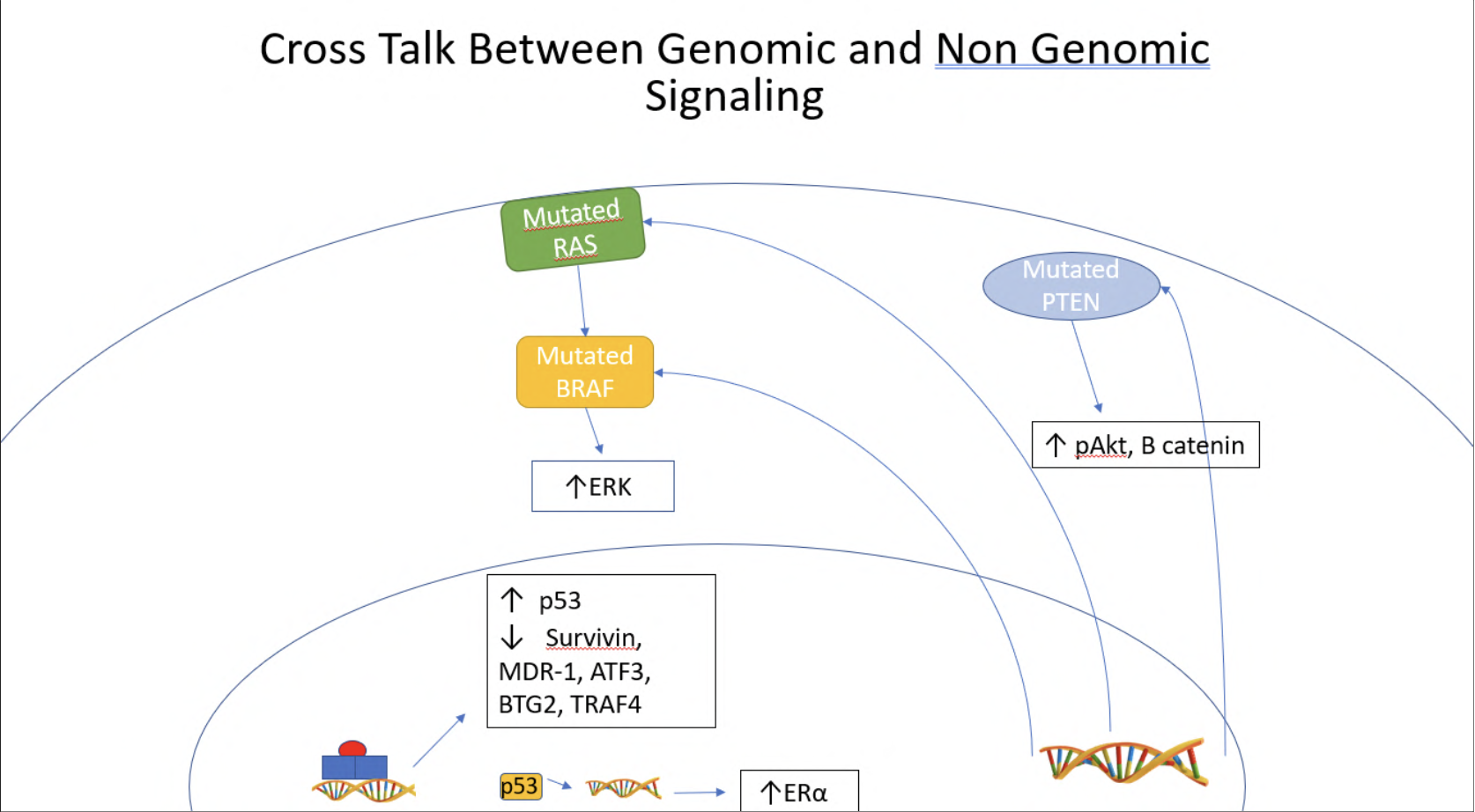
P53 serves as a "guardian" for the cellular genome, arresting the cell cycle, promoting DNA repair, and when necessary, promoting apoptosis. A large percentage of high grade serous and endometrioid carcinomas have shown loss of function mutations in p53, a gateway to carcinogenesis (38, 39). P53 has been shown to crosstalk with ER, forming a complex interaction between the two. ER α has been found to increase the transcription of the p53 gene within MCF7 breast cancer cells lines, both through a ligand dependent binding to an ERE in the p53 promoter (40) and through a calmodulin kinase IV mediated activation of NF-kB/CTF-1 transcription complex (41). P53 and its downstream mediator p21 have both been found to be upregulated with estradiol treatment within RhOSE ovarian cells as well (42). Furthermore, a polymorphism of p73, itself a homolog of p53 involved in the apoptosis of germ cells, has been found to be positively associated with increased ER positive human ovarian cancer development; this study surveyed epithelial, germ cell, and sex cord stromal cell tumors from human patients (43). However, ER α also plays an inhibitory role in regard to p53 regulated repression of survivin and MDR-1 (44) as well as p53 activation of ATF3, BTG2, and TRAF4 (45) as seen in Figure 2.
Wild type, non-mutated, p53 has been shown to increase the transcription of ER (46). In epithelial ovarian cancer, a mutated p53 protein that stabilizes wild type p53 was shown to increase levels of the ESR1 gene (47). It is hypothesized that the increased ESR1 expression is due to increased p53 mediated transcription of the ESR1 gene itself (47). However, owing to its anti-proliferative mechanism, a lack of p53 in mouse ovarian surface epithelial tumor cells correlated with estradiol mediated upregulation of growth and tumor invasion through upregulation of the estrogen receptor ESR1 (48). P53 and the estrogen receptor have a complex relationship, each increasing the transcription of the other while simultaneously inhibiting the transcriptional activity of that same molecule, preventing its downstream effects. A similar paradox can be seen in type 2 diabetes mellitus induced insulin resistance, in which high glucose levels drives increased insulin secretion but this subsequently results in resistance to insulin's effects.
Non-genomic activities of ER
Non genomic activities include those in which the estrogen receptor interacts with proteins other than transcription factors and do not immediately affect the genome. Ultimately many of these pathways lead to genomic alterations in the form of upregulation/downregulation of various genes, but begin independently of the genome. Table 3 details some of the therapies currently being studied, targeting these pathways.
Table 3. Summary of Treatments Targeting Non-Genomic Pathways
| Agent |
Mechanism of action |
Studies Performed |
References |
| Trastuzumab |
Her2 receptor targeting antibody |
Phase II study - 41 patients with primary peritoneal/recurrent or refractory ovarian carcinoma; 11.4% overexpression of Her2; response rate low at 7% |
Bookman, 83 |
| Trastuzumab/Pertuzumab |
Both Her2 receptor targeting antibodies |
Case report - high grade serous epithelial carcinoma stage IV; focal amplification of Her2 gene found; treated with combination trastuzumab/pertuzumab with persistent partial response of 37 months |
Thouvenin, 84 |
| Pertuzumab |
Her2 receptor targeting antibody |
Phase III study - 156 patients with platinum resistant ovarian carcinoma randomized to topotecan, gemcitabine, or paclitaxel monotherapy followed by adjuvant pertuzumab; no significant improvement in PFS/overall survival |
Lorusso, 85 |
| Gefitinib |
EGFR targeting antibody |
Phase Ib/II study - 19 patients with platinum resistant ovarian carcinoma, expressing EGFR, treated with topotecan and gefitinib; 16% had stable disease, 11% had partial response |
Chelariu-Raicu, 86 |
| Cetuximab |
EGFR targeting antibody |
Phase II study - 25 patients selected for single agent treatment with cetuximab; 1 patient achieved partial response, 9 achieved stable disease |
Schilder, 87 |
| Trametinib |
MEK inhibitor, targeting the MAPK pathway |
Phase II/III study - 260 patients with low grade serous carcinoma selected; 101 progression free survival events in treatment group vs 116 in control group; median progression free survival 13 months in treatment group vs 7.2 months in control |
Gershenson, 88 |
MAPK pathway and ER
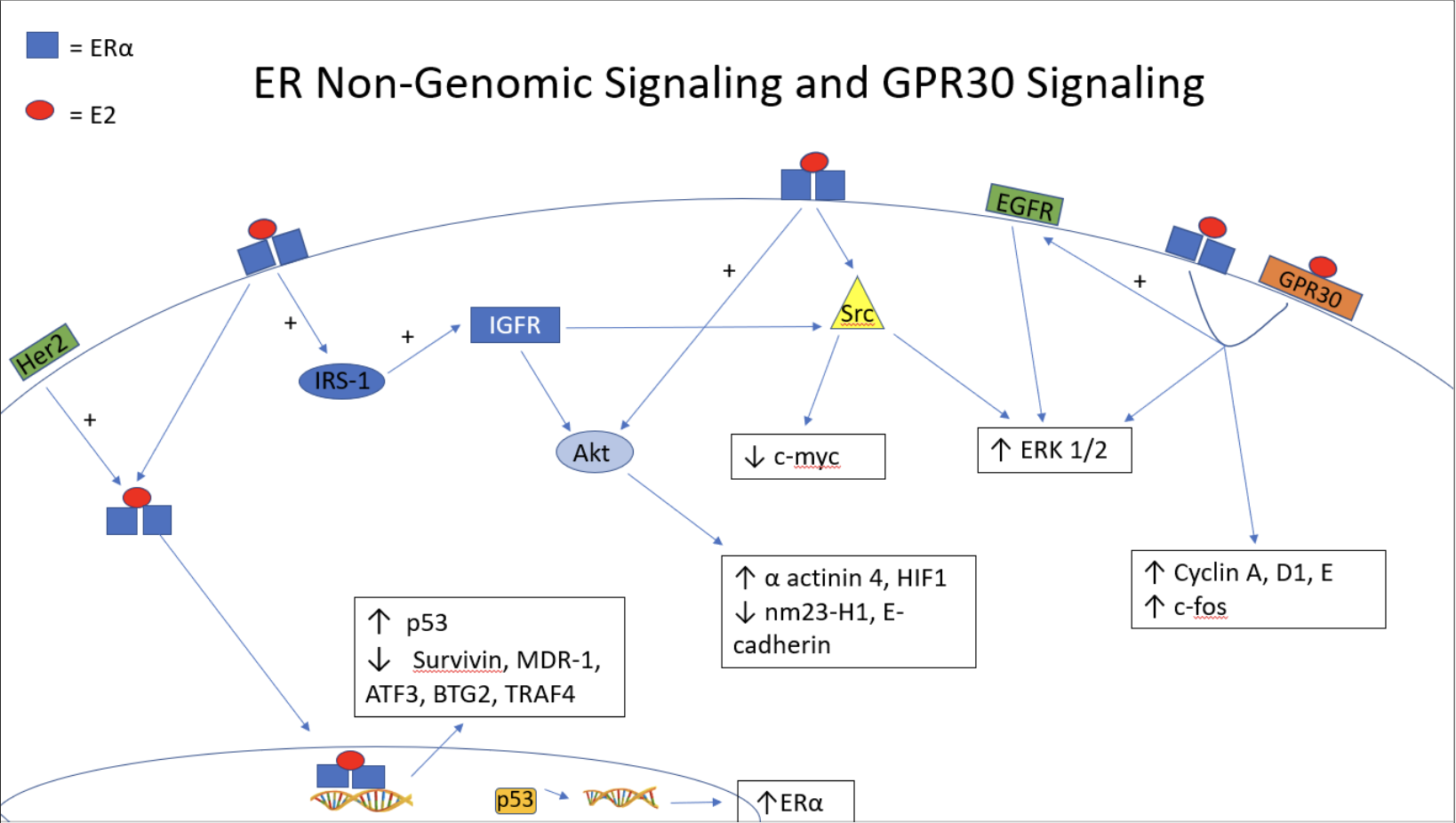
Mutations in both RAS and BRAF are present in higher frequency in low grade serous and mucinous ovarian carcinomas, upregulating the MAPK pathway, illustrated in Figure 2 (49, 50). Rap1A, a RAS associated protein, has been found to increase cell proliferation, migration, and invasion in human HEYA8 ovarian cancer cells. This carcinogenic activity appeared to be mediated by induced expression of MAPK pathways constituents, MEK1 and 2, as well as ERK 1 and 2, illustrated in Figure 3 (51). Furthermore, high amounts of phosphorylated/active MAPK have been observed in high grade serous carcinomas, corresponding to a worse survival rate (52). This increased level of MAPK correlated with a higher level of EGFR as well (52). Inhibition of the MAPK pathway, through the use of Aloesin (53) and delphinidin (54), has actually been shown to decrease the growth of SKOV3 ovarian cancer cells. The inhibition of Src kinase, which acts as a key regulator of both the MAPK and EGFR mediated pathways, has been investigated as well. Src was found to be highly expressed in ovarian cancer cell lines activated by the estrogen/ER complex (55). Subsequently, inhibition of Src inhibited cancer growth and reduced levels of c-myc expression within PEO1R and BG-1 ovarian cancer cells (55) seen in Figure 3.
EGFR/Her2 and ER
Her2/neu is a growth factor receptor commonly associated with carcinogenesis and has been shown to bind membrane associated ER, modulating its ligand responsive properties (56, 57). Her2 interacts with the MAPK pathway and has been shown to activate both nuclear ER and its coactivator AIB1 in order to increase ER mediated transcription (58). Therefore, the presence of Her2/neu in various carcinomas may enhance estrogen's role in carcinogenesis and tumor progression. High levels of Her2 have been reported in ovarian cancer, which reportedly caused increased stimulation of both MAPK and PI3K pathways. However, when inhibited by pertuzumab (a monoclonal anti-Her2 dimerization antibody), Her2 and ER α mediated activity is inhibited (59). Furthermore, EGFR has been found to be more highly expressed in ovarian cancer tissue when compared to normal ovarian tissue, with a 56.8% difference between the two groups (60). Higher EGFR expression in malignant ovarian tissue corresponds to a worse survival rate, owing to the anti-apoptotic effects of EGFR (61). As shown in Figure 3, EGFR gain of function mutations were found to promote increased phosphorylation of both Akt and ERK in ovarian cancer (62), ERK playing a role in the estrogen responsive MAPK pathway already mentioned above.
PI3K and ER
Lower grade endometrioid as well as clear cell carcinomas of the ovary showed higher rates of PTEN and PI3K pathway mutations, leading to an upregulation of this pathway and carcinogenesis (49, 39). In cases of low grade carcinoma, CTNNB1 (16-38% of cases) and PTEN (14-21% of cases) mutations result in increased activity of the B-catenin protein, causing unregulated cell proliferation through the PI3K cascade illustrated in Figure 2. However, in cases of high grade endometrioid carcinoma, mutations in P53 were found to be more prevalent (60% of cases), ultimately leading to unregulated DNA replication and cell proliferation (39). Shown in Figure 3, downstream of PI3K, Akt/pAkt and mTOR/pmTOR were shown to be phosphorylated in 55% of ovarian cancer tissue regulating transcription of Bcl-2 and survivin (63, 64). In ES-2 and SKOV-3 ovarian cancer cells, estradiol was found to reduce the expression of the nm23-H1 tumor suppressor gene (65). This inhibition of nm23-H1 expression was found to be mediated by pAkt, pointing to a PI3K/Akt mediated pathway in ovarian carcinogenesis (65). Furthermore, α-actinin 4, a metastatic promoter, and E-cadherin, a tumor suppressor gene, are both regulated by the PI3K pathway (fig 3). In SKOV3 cells, positive for both the α and β receptor, treatment with estradiol caused an increase in pAkt and increased α-actinin 4 expression, with concurrent decreased E-cadherin expression and increase in growth and migration (66). Another tumorigenic promoter, HIF-1α, expressed under low oxygen conditions, was found to be elevated post-estradiol treatment in ES-2 and SKOV3 cells, promoting cell proliferation. This elevation was found to be dependent on estradiol's activation of Akt (67).
IGF/IGFR and ER
IGF is involved in normal function and development of many tissues including the ovary. Given its role in steroidogenesis and overall cell growth, it has been thought that IGF-I and IGF-II, as well as their receptors and the related 6 IGF binding proteins (IGFBP), might play a role in carcinogenesis. IGF-I has been found to facilitate proliferation, invasion and angiogenesis in tumor cells and IGF-II has been shown to mediate cell adhesion and invasion in ovarian cancer (68). IGFBPs play an inhibitory role in normal physiologic development, acting as a trap for IGF-I and IGF-II (69), but in ovarian cancer, a downregulation of IGFBP3 and 5 with a concurrent increase in IGFBP4 (70) and IGFBP2 (71) levels has been observed. The mechanism behind this selective regulation of IGF binding proteins is unknown but has been suggested to be a result of ER α influence (72). Furthermore, as shown in Figure 3, the IGF-I receptor has been found to upregulate both the MAPK and PI3K pathways mentioned above (73-76), as well as downstream constituents Akt and ERK1 and ERK2 (68). This signaling generally occurs through the phosphorylation of the insulin receptor substrate-1 (IRS-1), which has been shown to be activated by the estradiol/ER α complex (73).
GPR30 Receptor
The G protein coupled receptor is referred to as either GPR30 or GPER1 and both labels will be used interchangeably in the subsequent section. The ligands to which GPR30 responds include estradiol, G-1 (G protein coupled receptor agonist), as well as tamoxifen and other estrogen agonists (77). GPR30 has been found to localize primarily in 2 locations: the plasma membrane and the endoplasmic reticulum (78, 77), with estrogen responsivity at both locations. Ultimately, GPR30 exerts its effects in ovarian cancer through calcium mobilization (77) and EGFR transactivation with subsequent MAPK activation (79,80). GPR30 expression is higher in ovarian carcinomas, as opposed to benign and borderline malignancies, with its highest expression in serous, endometrioid and mucinous tumors (80). In BG-1 ovarian cancer cells, EGFR/ERK activation was found to be dependent on both ER α and GPR30, effectively promoting cell proliferation (79). Furthermore, estradiol was found to upregulate cyclins A, E, and D1 as well as the c-fos gene, through this same ER α/GPR30 mechanism, further promoting proliferation and cell cycle progression (79), shown in Figure 3. GPR30/EGFR co-expression in ovarian carcinomas has been shown to correlate with worse survival rates in these tumors (80). Paradoxically, GPER1 has been found to be highly expressed in granulosa cell tumors and was found to be associated with reduced migration and invasion in these tumors specifically. This reduced migratory/invasive property was shown to be mediated by GPER1 and repression of ERK1 and ERK2 (81).
Conclusion
Estrogen has been linked to breast, endometrial, and ovarian cancer development. Its involvement in the pathogenesis of ovarian cancer is complicated, given the presence of ER α and β, as well as GPR30 and the individual mechanisms by which each acts. ER-α has been shown to be primarily carcinogenic while ER-β is a known negative regulator of carcinogenesis; GPR30 is primarily carcinogenic but there is evidence to suggest it could be a negative regulator of the cell cycle as well in ovarian tissue. Furthermore, these receptors interact with the p53, MAPK, EGFR/Her2, PI3K, and IGF/IGFR pathways, with unique interactions with each pathway. Estrogen and its effects in ovarian cancer are still poorly characterized, particularly regarding these molecular signaling pathways, an area of research that may yield unique insights into treatment of this disease.
Disclosures
Conflicts of interest: None.
Availability of data and material: Not applicable.
Code availability: Not applicable.
Authors' contributions: Authors listed in the manuscript have contributed per submission guidelines and standards for authorship.
Ethics approval: Not applicable.
Consent to participate: Not applicable.
References
- Siegel, R.L., Miller, K.D. and Jemal, A. (2020). Cancer statistics, 2020. CA: a cancer journal for clinicians, 70(1), 7–30.
- Ehmann T, Barel S, Xu G, et al. (2019). A comparative analysis of estrogenic pathways between ovarian and cervical cancers. Proceedings of the LECOM Inter-Professional Research Day (Erie, PA), A7.
- Orr, B. and Edwards, R.P. (2018). Diagnosis and treatment of ovarian cancer. Hematology/Oncology Clinics, 32(6), 943-964.
- Howlader N, Noone AM, Krapcho M, et al. (2020). SEER Cancer Statistics Review, 1975-2017 [Online]. Available from: https://seer.cancer.gov/archive/csr/1975_2017/. [Accessed Aug 1 2021]
- Simpkins, F., Garcia-Soto, A. and Slingerland, J. (2013). New insights on the role of hormonal therapy in ovarian cancer. Steroids, 78(6), 530-537.
- Flouriot, G., Brand, H., Denger, S., et al. (2000). Identification of a new isoform of the human estrogen receptor-alpha (hER-α) that is encoded by distinct transcripts and that is able to repress hER-α activation function 1. The EMBO journal, 19(17), 4688-4700.
- Shi, L., Dong, B., Li, Z., et al. (2009). Expression of ER-α36, a novel variant of estrogen receptor α, and resistance to tamoxifen treatment in breast cancer. Journal of clinical Oncology, 27(21), 3423.
- Sieh, W., Köbel, M., Longacre, T.A., et al. (2013). Hormone-receptor expression and ovarian cancer survival: an Ovarian Tumor Tissue Analysis consortium study. The lancet oncology, 14(9), 853-862.
- Pujol, P., Rey, J.M., Nirde, P., et al. (1998). Differential expression of estrogen receptor-α and-β messenger RNAs as a potential marker of ovarian carcinogenesis. Cancer research, 58(23), 5367-5373.
- O'Donnell, A.J., Macleod, K.G., Burns, D.J., et al. (2005). Estrogen receptor-α mediates gene expression changes and growth response in ovarian cancer cells exposed to estrogen. Endocrine-related cancer, 12(4), 851-866.
- Langdon, S.P., Hawkes, M.M., Lawrie, S.S., et al. (1990). Oestrogen receptor expression and the effects of oestrogen and tamoxifen on the growth of human ovarian carcinoma cell lines. British Journal of Cancer, 62(2), 213-216.
- Voutsadakis, I.A. (2016). Hormone receptors in serous ovarian carcinoma: prognosis, pathogenesis, and treatment considerations. Clinical Medicine Insights: Oncology, 10, CMO-S32813.
- Herynk, M.H. and Fuqua, S.A. (2004). Estrogen receptor mutations in human disease. Endocrine reviews, 25(6), 869-898.
- Bossard, C., Busson, M., Vindrieux, D., et al. (2012). Potential Role of Estrogen Receptor Beta as a Tumor Suppressor of Epithelial Ovarian Cancer. PLoS ONE, 7(9), e44787.
- Treeck, O., Pfeiler, G., Mitter, D., et al. (2007). Estrogen receptor β1 exerts antitumoral effects on SK-OV-3 ovarian cancer cells. Journal of Endocrinology, 193(3), 421-433.
- Prossnitz, E.R. and Barton, M. (2014). Estrogen biology: new insights into GPER function and clinical opportunities. Molecular and cellular endocrinology, 389(1-2), 71-83.
- Albanito, L., Madeo, A., Lappano, R., et al. (2007). G protein–coupled receptor 30 (GPR30) mediates gene expression changes and growth response to 17β-estradiol and selective GPR30 ligand G-1 in ovarian cancer cells. Cancer research, 67(4), 1859-1866.
- Long, L., Cao, Y. and Tang, L.D. (2012). Transmembrane estrogen receptor GPR30 is more frequently expressed in malignant than benign ovarian endometriotic cysts and correlates with MMP-9 expression. International Journal of Gynecologic Cancer, 22(4).
- Smith, H.O., Arias-Pulido, H., Kuo, D.Y., et al. (2009). GPR30 predicts poor survival for ovarian cancer. Gynecologic oncology, 114(3), 465-471.
- Heublein, S., Mayr, D., Friese, K., et al. (2014). The G-protein-coupled estrogen receptor (GPER/GPR30) in ovarian granulosa cell tumors. International journal of molecular sciences, 15(9), 15161-15172.
- Kolkova, Z., Casslén, V., Henic, E., et al. (2012). The G protein-coupled estrogen receptor 1 (GPER/GPR30) does not predict survival in patients with ovarian cancer. Journal of ovarian research, 5(1), 1-11.
- Ignatov, T., Modl, S., Thulig, M., et al. (2013). GPER-1 acts as a tumor suppressor in ovarian cancer. Journal of ovarian research, 6(1), 1-10.
- Katzenellenbogen, B.S., (1996). Estrogen receptors: bioactivities and interactions with cell signaling pathways. Biology of reproduction, 54(2), 287-293.
- O'Malley, B.W. (2005). A life-long search for the molecular pathways of steroid hormone action. Molecular Endocrinology, 19(6), 1402-1411.
- Weigel, N.L. (1996). Steroid hormone receptors and their regulation by phosphorylation. Biochemical Journal, 319(3), 657-667.
- Pietras, R.J. and Márquez-Garbán, D.C. (2007). Membrane-associated estrogen receptor signaling pathways in human cancers. Clinical Cancer Research, 13(16), 4672-4676.
- Razandi, M., Pedram, A., Greene, G.L. et al. (1999). Cell membrane and nuclear estrogen receptors (ERs) originate from a single transcript: studies of ERα and ERβ expressed in Chinese hamster ovary cells. Molecular endocrinology, 13(2), 307-319.
- Bjornstrom, L. and Sjoberg, M. (2005). Mechanisms of estrogen receptor signaling: convergence of genomic and nongenomic actions on target genes. Molecular endocrinology, 19(4), 833-842.
- Hall, J.M. and Korach, K.S. (2003). Stromal cell-derived factor 1, a novel target of estrogen receptor action, mediates the mitogenic effects of estradiol in ovarian and breast cancer cells. Molecular Endocrinology, 17(5), 792-803.
- Webb, P., Nguyen, P., Valentine, C., et al. (1999). The estrogen receptor enhances AP-1 activity by two distinct mechanisms with different requirements for receptor transactivation functions. Molecular Endocrinology, 13(10), 1672-1685.
- Anzick, S.L., Kononen, J., Walker, R.L., et al. (1997). AIB1, a steroid receptor coactivator amplified in breast and ovarian cancer. Science, 277(5328), 965-968.
- Liang, M. and Zhao, J. (2018). Protein expressions of AIB1, p53 and Bcl-2 in epithelial ovarian cancer and their correlations with the clinical pathological features and prognosis. Eur Rev Med Pharmacol Sci, 22(16), 5134-5139.
- Palmieri, C., Gojis, O., Rudraraju, B., et al. (2013). Expression of steroid receptor coactivator 3 in ovarian epithelial cancer is a poor prognostic factor and a marker for platinum resistance. British journal of cancer, 108(10), 2039-2044.
- Jakacka, M., Ito, M., Weiss, J, et al. (2001). Estrogen receptor binding to DNA is not required for its activity through the nonclassical AP1 pathway. Journal of Biological Chemistry, 276(17), 13615-13621.
- Sasaki, H., Hayakawa, J., Terai, Y., et al. (2008). Difference between genomic actions of estrogen versus raloxifene in human ovarian cancer cell lines. Oncogene, 27(19), 2737-2745.
- Hein, S., Mahner, S., Kanowski, C., et al. (2009). Expression of Jun and Fos proteins in ovarian tumors of different malignant potential and in ovarian cancer cell lines. Oncology reports, 22(1), 177-183.
- Bardin, A., Moll, F., Margueron, R., et al. (2005). Transcriptional and posttranscriptional regulation of fibulin-1 by estrogens leads to differential induction of messenger ribonucleic acid variants in ovarian and breast cancer cells. Endocrinology, 146(2), 760-768.
- Cotran, R., Kumar, V. and Robbins, S. (2015). Pathologic basis of disease. Philadelphia, PA: Saunders Elsevier.
- Cho, K.R. and Shih, I.M. (2009). Ovarian cancer. Annual review of pathology: mechanisms of disease, 4, 287-313.
- Berger, C.E., Qian, Y., Liu, G., et al. (2012). p53, a target of estrogen receptor (ER) α, modulates DNA damage-induced growth suppression in ER-positive breast cancer cells. Journal of Biological Chemistry, 287(36), 30117-30127.
- Qin, C., Nguyen, T., Stewart, J., et al. (2002). Estrogen up-regulation of p53 gene expression in MCF-7 breast cancer cells is mediated by calmodulin kinase IV-dependent activation of a nuclear factor κB/CCAAT-binding transcription factor-1 complex. Molecular Endocrinology, 16(8), 1793-1809.
- Wright, J.W., Stouffer, R.L. and Rodland, K.D. (2005). High-dose estrogen and clinical selective estrogen receptor modulators induce growth arrest, p21, and p53 in primate ovarian surface epithelial cells. The Journal of Clinical Endocrinology & Metabolism, 90(6), 3688-3695.
- Guan, X., Zhang, N., Yin, Y., et al. (2012). Polymorphisms in the p63 and p73 genes are associated with ovarian cancer risk and clinicopathological variables. Journal of Experimental & Clinical Cancer Research, 31(1), 1-8.
- Sayeed, A., Konduri, S.D., Liu, W., et al. (2007). Estrogen receptor α inhibits p53-mediated transcriptional repression: implications for the regulation of apoptosis. Cancer research, 67(16), 7746-7755.
- Bailey, S.T., Shin, H., Westerling, T., et al. (2012). Estrogen receptor prevents p53-dependent apoptosis in breast cancer. Proceedings of the National Academy of Sciences, 109(44), 18060-18065.
- Berger, C., Qian, Y. and Chen, X. (2013). The p53-estrogen receptor loop in cancer. Current molecular medicine, 13(8), 1229-1240.
- Ren, Y.A., Mullany, L.K., Liu, Z., et al. (2016). Mutant p53 promotes epithelial ovarian cancer by regulating tumor differentiation, metastasis, and responsiveness to steroid hormones. Cancer research, 76(8), 2206-2218.
- Mullany, L.K., Liu, Z., Wong, K.K., et al. (2014). Tumor repressor protein 53 and steroid hormones provide a new paradigm for ovarian cancer metastases. Molecular Endocrinology, 28(1), 127-137.
- Rosen, D.G., Yang, G., Liu, G., et al. (2009). Ovarian cancer: pathology, biology, and disease models. Frontiers in bioscience: a journal and virtual library, 14, 2089.
- Ho, C.L., Kurman, R.J., Dehari, R., et al. (2004). Mutations of BRAF and KRAS precede the development of ovarian serous borderline tumors. Cancer research, 64(19), 6915-6918.
- Lu, L., Wang, J., Wu, Y., et al. (2016). Rap1A promotes ovarian cancer metastasis via activation of ERK/p38 and notch signaling. Cancer medicine, 5(12), 3544-3554.
- Hew, K.E., Miller, P.C., El-Ashry, D., et al. (2016). MAPK activation predicts poor outcome and the MEK inhibitor, selumetinib, reverses antiestrogen resistance in ER-positive high-grade serous ovarian cancer. Clinical Cancer Research, 22(4), 935-947.
- Zhang, L.Q., Lv, R.W., Qu, X.D., et al. (2017). Aloesin suppresses cell growth and metastasis in ovarian cancer SKOV3 cells through the inhibition of the MAPK signaling pathway. Analytical Cellular Pathology, 2017, 8158254.
- Lim, W. and Song, G. (2017). Inhibitory effects of delphinidin on the proliferation of ovarian cancer cells via PI3K/AKT and ERK 1/2 MAPK signal transduction. Oncology Letters, 14(1), 810-818.
- Simpkins, F., Hevia-Paez, P., Sun, J., et al. (2012). Src Inhibition with saracatinib reverses fulvestrant resistance in ER-positive ovarian cancer models in vitro and in vivo. Clinical cancer research, 18(21), 5911-5923.
- Chung, Y.L., Sheu, M.L., Yang, S.C., et al. (2002). Resistance to tamoxifen‐induced apoptosis is associated with direct interaction between Her2/neu and cell membrane estrogen receptor in breast cancer. International journal of cancer, 97(3), 306-312.
- Arpino, G., Wiechmann, L., Osborne, C.K. et al. (2008). Crosstalk between the estrogen receptor and the HER tyrosine kinase receptor family: molecular mechanism and clinical implications for endocrine therapy resistance. Endocrine reviews, 29(2), 217-233.
- Osborne, C.K., Bardou, V., Hopp, T.A., et al. (2003). Role of the estrogen receptor coactivator AIB1 (SRC-3) and HER-2/neu in tamoxifen resistance in breast cancer. Journal of the National Cancer Institute, 95(5), 353-361.
- Mullen, P., Cameron, D.A., Hasmann, M., et al. (2007). Sensitivity to pertuzumab (2C4) in ovarian cancer models: cross-talk with estrogen receptor signaling. Molecular cancer therapeutics, 6(1), 93-100.
- Ranjbar, R., Nejatollahi, F., Ahmadi, A.S.N., et al. (2015). Expression of vascular endothelial growth factor (VEGF) and epidermal growth factor receptor (EGFR) in patients with serous ovarian carcinoma and their clinical significance. Iranian journal of cancer prevention, 8(4).
- Psyrri, A., Kassar, M., Yu, Z., et al. (2005). Effect of epidermal growth factor receptor expression level on survival in patients with epithelial ovarian cancer. Clinical Cancer Research, 11(24), 8637-8643.
- Tanaka, Y., Terai, Y., Tanabe, A., et al. (2011). Prognostic effect of epidermal growth factor receptor gene mutations and the aberrant phosphorylation of Akt and ERK in ovarian cancer. Cancer biology & therapy, 11(1), 50-57.
- Altomare, D.A., Wang, H.Q., Skele, K.L., et al. (2004). AKT and mTOR phosphorylation is frequently detected in ovarian cancer and can be targeted to disrupt ovarian tumor cell growth. Oncogene, 23(34), 5853-5857.
- Cheaib, B., Auguste, A. and Leary, A. (2015). The PI3K/Akt/mTOR pathway in ovarian cancer: therapeutic opportunities and challenges. Chinese journal of cancer, 34(1), 4-16.
- Hua, K., Feng, W., Cao, Q., et al. (2008). Estrogen and progestin regulate metastasis through the PI3K/AKT pathway in human ovarian cancer. International journal of oncology, 33(5), 959-967.
- Lu, Z., Zhang, Y., Yan, X., et al. (2014). Estrogen stimulates the invasion of ovarian cancer cells via activation of the PI3K/AKT pathway and regulation of its downstream targets E-cadherin and α-actinin-4. Molecular Medicine Reports, 10(5), 2433-2440.
- Hua, K., Din, J., Cao, Q., et al. (2009). Estrogen and progestin regulate HIF-1α expression in ovarian cancer cell lines via the activation of Akt signaling transduction pathway. Oncology reports, 21(4), 893-898.
- Beauchamp, M.C., Yasmeen, A., Knafo, A. et al. (2010). Targeting insulin and insulin-like growth factor pathways in epithelial ovarian cancer. Journal of oncology, 2010, 257058.
- Foulstone, E., Prince, S., Zaccheo, O., et al. (2005). Insulin‐like growth factor ligands, receptors, and binding proteins in cancer. The Journal of Pathology: A Journal of the Pathological Society of Great Britain and Ireland, 205(2), 145-153.
- Walker, G., MacLeod, K., Williams, A.R., et al. (2007). Insulin-like growth factor binding proteins IGFBP3, IGFBP4, and IGFBP5 predict endocrine responsiveness in patients with ovarian cancer. Clinical Cancer Research, 13(5), 1438-1444.
- Flyvbjerg, A., Mogensen, O., Mogensen, B., et al. (1997). Elevated serum insulin-like growth factor-binding protein 2 (IGFBP-2) and decreased IGFBP-3 in epithelial ovarian cancer: correlation with cancer antigen 125 and tumor-associated trypsin inhibitor. The Journal of Clinical Endocrinology & Metabolism, 82(7), 2308-2313.
- Lu, L., Katsaros, D., Wiley, A., et al. (2006). The relationship of insulin-like growth factor-II, insulin-like growth factor binding protein-3, and estrogen receptor-alpha expression to disease progression in epithelial ovarian cancer. Clinical Cancer Research, 12(4), 1208-1214.
- Kahlert, S., Nuedling, S., Van Eickels, M., et al. (2000). Estrogen receptor α rapidly activates the IGF-1 receptor pathway. Journal of Biological Chemistry, 275(24), 18447-18453.
- Chen, J., Zhao, K.N., Li, R., et al. (2014). Activation of PI3K/Akt/mTOR pathway and dual inhibitors of PI3K and mTOR in endometrial cancer. Current medicinal chemistry, 21(26), 3070-3080.
- Fukushima, T., Nakamura, Y., Yamanaka, D., et al. (2012). Phosphatidylinositol 3-kinase (PI3K) activity bound to insulin-like growth factor-I (IGF-I) receptor, which is continuously sustained by IGF-I stimulation, is required for IGF-I-induced cell proliferation. Journal of Biological Chemistry, 287(35), 29713-29721.
- Cao, Z., Liu, L.Z., Dixon, D.A., et al. (2007). Insulin-like growth factor-I induces cyclooxygenase-2 expression via PI3K, MAPK and PKC signaling pathways in human ovarian cancer cells. Cellular signalling, 19(7), 1542-1553.
- Qian, H., Xuan, J., Liu, Y. et al. (2016). Function of G-protein-coupled estrogen receptor-1 in reproductive system tumors. Journal of immunology research, 2016, 7128702.
- Revankar, C.M., Cimino, D.F., Sklar, L.A., et al. (2005). A transmembrane intracellular estrogen receptor mediates rapid cell signaling. Science, 307(5715), 1625-1630.
- Albanito, L., Madeo, A., Lappano, R., et al. (2007). G protein–coupled receptor 30 (GPR30) mediates gene expression changes and growth response to 17β-estradiol and selective GPR30 ligand G-1 in ovarian cancer cells. Cancer research, 67(4), 1859-1866.
- Fujiwara, S., Terai, Y., Kawaguchi, H., et al. (2012). GPR30 regulates the EGFR-Akt cascade and predicts lower survival in patients with ovarian cancer. Journal of ovarian research, 5(1), 1-10.
- François, C.M., Wargnier, R., Petit, F., et al. (2015). 17β-estradiol inhibits spreading of metastatic cells from granulosa cell tumors through a non-genomic mechanism involving GPER1. Carcinogenesis, 36(5), 564-573.
- Schüler-Toprak, S., Weber, F., Skrzypczak, M., et al. (2018). Estrogen receptor β is associated with expression of cancer associated genes and survival in ovarian cancer. BMc cancer, 18(1), 1-9.
- Bookman, M.A., Darcy, K.M., Clarke-Pearson, D., et al. (2003). Evaluation of monoclonal humanized anti-HER2 antibody, trastuzumab, in patients with recurrent or refractory ovarian or primary peritoneal carcinoma with overexpression of HER2: a phase II trial of the Gynecologic Oncology Group. Journal of clinical oncology, 21(2), 283-290.
- Thouvenin, L., Charrier, M., Clement, S., et al. (2021). Ovarian cancer with high-level focal ERBB2 amplification responds to trastuzumab and pertuzumab. Gynecologic Oncology Reports, 37, 100787.
- Lorusso, D., Hilpert, F., Martin, A.G., et al. (2019). Patient-reported outcomes and final overall survival results from the randomized phase 3 PENELOPE trial evaluating pertuzumab in low tumor human epidermal growth factor receptor 3 (HER3) mRNA-expressing platinum-resistant ovarian cancer. International Journal of Gynecologic Cancer, 29(7).
- Chelariu-Raicu, A., Levenback, C.F., Slomovitz, B.M., et al. (2020). Phase Ib/II study of weekly topotecan and daily gefitinib in patients with platinum resistant ovarian, peritoneal, or fallopian tube cancer. International Journal of Gynecologic Cancer, 30(11).
- Schilder, R.J., Pathak, H.B., Lokshin, A.E., et al. (2009). Phase II trial of single agent cetuximab in patients with persistent or recurrent epithelial ovarian or primary peritoneal carcinoma with the potential for dose escalation to rash. Gynecologic oncology, 113(1), 21-27.
- Gershenson, D.M., Miller, A., Brady, W.E., et al. (2022). Trametinib versus standard of care in patients with recurrent low-grade serous ovarian cancer (GOG 281/LOGS): an international, randomised, open-label, multicentre, phase 2/3 trial. The Lancet, 399(10324), 541-553.
- He, Y., Alejo, S., Venkata, P.P., Johnson, J.D., Loeffel, I., Pratap, U.P., Zou, Y., Lai, Z., Tekmal, R.R., Kost, E.R. and Sareddy, G.R. (2022). Therapeutic targeting of ovarian Cancer stem cells using estrogen receptor Beta agonist. International journal of molecular sciences, 23(13), 7159.
- Banerjee, A., Cai, S., Xie, G., Li, N., Bai, X., Lavudi, K., Wang, K., Zhang, X., Zhang, J., Patnaik, S. and Backes, F.J. (2022). A Novel Estrogen Receptor β Agonist Diminishes Ovarian Cancer Stem Cells via Suppressing the Epithelial-to-Mesenchymal Transition. Cancers, 14(9), 2311.